
Microscopía de súper resolución - Super-resolution microscopy
La microscopía de superresolución es una serie de técnicas en microscopía óptica que permiten que dichas imágenes tengan resoluciones superiores a las impuestas por el límite de difracción , que se debe a la difracción de la luz . Las técnicas de obtención de imágenes de superresolución se basan en el campo cercano (microscopía de túnel de fotones, así como en las que utilizan Pendry Superlens y microscopía óptica de barrido de campo cercano ) o en el campo lejano . Entre las técnicas que se basan en este último se encuentran aquellas que mejoran la resolución solo modestamente (hasta aproximadamente un factor de dos) más allá del límite de difracción, como la microscopía confocal con orificio de alfiler cerrado o con la ayuda de métodos computacionales como la deconvolución o el píxel basado en detector. reasignación (por ejemplo, microscopía de nuevo escaneo, reasignación de píxeles), el microscopio 4Pi y tecnologías de microscopía de iluminación estructurada como SIM y SMI .
Hay dos grupos principales de métodos para microscopía de superresolución en el campo lejano que pueden mejorar la resolución en un factor mucho mayor:
- Superresolución determinista: los emisores más utilizados en microscopía biológica, los fluoróforos , muestran una respuesta no lineal a la excitación, que puede aprovecharse para mejorar la resolución. Dichos métodos incluyen STED , GSD , RESOLFT y SSIM.
- Superresolución estocástica: la complejidad química de muchas fuentes de luz molecular les da un comportamiento temporal complejo, que puede usarse para hacer que varios fluoróforos cercanos emitan luz en momentos separados y, por lo tanto, se puedan resolver en el tiempo. Estos métodos incluyen imágenes de fluctuación óptica de súper resolución (SOFI) y todos los métodos de localización de una sola molécula (SMLM), como SPDM , SPDMphymod , PALM , FPALM, STORM y dSTORM.
El 8 de octubre de 2014, se otorgó el Premio Nobel de Química a Eric Betzig , WE Moerner y Stefan Hell por "el desarrollo de la microscopía de fluorescencia súper resuelta ", que lleva " la microscopía óptica a la nanodimensión ". Las diferentes modalidades de microscopía de superresolución están siendo adoptadas cada vez más por la comunidad de investigación biomédica, y estas técnicas se están convirtiendo en herramientas indispensables para comprender la función biológica a nivel molecular.
Historia
En 1978, se habían desarrollado las primeras ideas teóricas para romper el límite de Abbe , que requería el uso de un microscopio 4Pi como microscopio de fluorescencia de escaneo láser confocal donde la luz se enfoca desde todos los lados hacia un foco común que se usa para escanear el objeto. por excitación 'punto por punto' combinada con detección 'punto por punto'. Sin embargo, la publicación de 1978 había sacado una conclusión física inadecuada (es decir, un punto de luz en forma de punto) y había pasado por alto por completo el aumento de la resolución axial como el beneficio real de agregar el otro lado del ángulo sólido.
Parte de la siguiente información se recopiló (con permiso) de la revisión de un blog de química de las técnicas de microscopía de sub-difracción.
En 1986, Okhonin patentó un microscopio óptico de superresolución basado en emisión estimulada.
Técnicas de superresolución
Microscopía de túnel de fotones (PTM)
Mejora local / ANSOM / nano antenas ópticas
Microscopía de mapeo óptico aleatorio de campo cercano (NORM)
La microscopía de mapeo óptico aleatorio de campo cercano (NORM) es un método de adquisición óptica de campo cercano mediante un microscopio de campo lejano a través de la observación del movimiento browniano de las nanopartículas en un líquido de inmersión.
NORM utiliza el escaneo de la superficie del objeto mediante nanopartículas que se mueven estocásticamente. A través del microscopio, las nanopartículas parecen puntos redondos simétricos. El ancho del punto es equivalente a la función de dispersión del punto (~ 250 nm) y está definido por la resolución del microscopio. Las coordenadas laterales de la partícula dada se pueden evaluar con una precisión mucho mayor que la resolución del microscopio. Al recopilar la información de muchos marcos, se puede trazar la distribución de la intensidad del campo cercano en todo el campo de visión del microscopio. En comparación con NSOM y ANSOM, este método no requiere ningún equipo especial para el posicionamiento de la punta y tiene un gran campo de visión y una profundidad de enfoque. Debido a la gran cantidad de "sensores" de escaneo, se puede lograr la adquisición de imágenes en un tiempo más corto.
4Pi
Un microscopio 4Pi es un microscopio de fluorescencia de escaneo láser con una resolución axial mejorada . El valor típico de 500 a 700 nm se puede mejorar a 100 a 150 nm, lo que corresponde a un punto focal casi esférico con 5 a 7 veces menos volumen que el de la microscopía confocal estándar .
La mejora en la resolución se logra mediante el uso de dos lentes de objetivo opuestos, ambos enfocados en la misma ubicación geométrica. Además, la diferencia en la longitud del camino óptico a través de cada uno de los dos objetivos se minimiza cuidadosamente. De este modo, las moléculas que residen en el área focal común de ambos objetivos pueden iluminarse coherentemente desde ambos lados, y la luz reflejada o emitida puede recogerse coherentemente, es decir, es posible una superposición coherente de la luz emitida en el detector. El ángulo sólido que se utiliza para la iluminación y la detección aumenta y se acerca al caso ideal, donde la muestra se ilumina y detecta desde todos los lados simultáneamente.

Hasta ahora, la mejor calidad en un microscopio 4Pi se ha alcanzado junto con la microscopía STED en células fijas y la microscopía RESOLFT con proteínas intercambiables en células vivas.
Microscopía de iluminación estructurada (SIM)

La microscopía de iluminación estructurada (SIM) mejora la resolución espacial al recopilar información del espacio de frecuencias fuera de la región observable. Este proceso se realiza en el espacio recíproco: la transformada de Fourier (FT) de una imagen SI contiene información adicional superpuesta de diferentes áreas del espacio recíproco; con varios fotogramas donde la iluminación se desplaza en alguna fase , es posible separar y reconstruir computacionalmente la imagen FT, que tiene mucha más información de resolución. El FT inverso devuelve la imagen reconstruida a una imagen de superresolución.
La microscopía SIM podría potencialmente reemplazar la microscopía electrónica como herramienta para algunos diagnósticos médicos. Estos incluyen el diagnóstico de trastornos renales, cáncer de riñón y enfermedades de la sangre.
Aunque el término "microscopía de iluminación estructurada" fue acuñado por otros en años posteriores, Guerra (1995) publicó por primera vez resultados en los que la luz modelada por una rejilla de tono de 50 nm iluminaba una segunda rejilla de tono de 50 nm, con las rejillas giradas con respecto a cada una de ellas. otros por la cantidad angular necesaria para lograr el aumento. Aunque la longitud de onda de iluminación era de 650 nm, la rejilla de 50 nm se resolvió fácilmente. Esto mostró una mejora de casi 5 veces sobre el límite de resolución de Abbe de 232 nm que debería haber sido el más pequeño obtenido para la apertura numérica y la longitud de onda utilizada. En un desarrollo posterior de este trabajo, Guerra demostró que la topografía lateral superesuelta se logra cambiando de fase el campo evanescente. Varias patentes estadounidenses se otorgaron a Guerra individualmente o con colegas, y se asignaron a Polaroid Corporation . Las licencias para esta tecnología fueron adquiridas por Dyer Energy Systems, Calimetrics Inc. y Nanoptek Corp. para el uso de esta técnica de superresolución en el almacenamiento de datos ópticos y microscopía.
- Imágenes de núcleos celulares y estadios mitóticos registradas con 3D-SIM.

Comparación de microscopía confocal - 3D-SIM
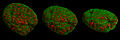
Núcleo celular en profase desde varios ángulos
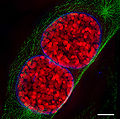
Dos núcleos de células de ratón en profase.

célula de ratón en telofase



Iluminación modulada espacialmente (SMI)

Una implementación de iluminación estructurada se conoce como iluminación modulada espacialmente (SMI). Al igual que la iluminación estructurada estándar, la técnica SMI modifica la función de dispersión de puntos (PSF) de un microscopio de una manera adecuada. En este caso, sin embargo, "la resolución óptica en sí no se mejora"; en cambio, la iluminación estructurada se utiliza para maximizar la precisión de las mediciones de distancia de los objetos fluorescentes, para "permitir mediciones de tamaño en dimensiones moleculares de unas pocas decenas de nanómetros".
El microscopio Vertico SMI logra una iluminación estructurada mediante el uso de uno o dos rayos láser de interferencia opuestos a lo largo del eje. A continuación, el objeto que se está fotografiando se mueve en pasos de alta precisión a través del campo de ondas, o el campo de ondas en sí mismo se mueve en relación con el objeto mediante cambios de fase. Esto da como resultado un tamaño axial mejorado y una resolución de distancia mejorada.
SMI se puede combinar con otras tecnologías de superresolución, por ejemplo con 3D LIMON o LSI- TIRF como un interferómetro de reflexión interna total con iluminación estructurada lateralmente (este último instrumento y técnica es esencialmente un microscopio de túnel de fotones de fase desplazada, que emplea un microscopio de luz de reflexión con campo evanescente de fase desplazada (Guerra, 1996)). Esta técnica SMI permite adquirir imágenes ópticas de luz de distribuciones de autofluoróforos en secciones de tejido del ojo humano con una resolución óptica sin precedentes. El uso de tres longitudes de onda de excitación diferentes (488, 568 y 647 nm) permite recopilar información espectral sobre la señal de autofluorescencia. Esto se ha utilizado para examinar el tejido del ojo humano afectado por la degeneración macular .
Técnicas funcionales deterministas
La microscopía de transiciones de fluorescencia óptica saturable reversible (RESOLFT) es una microscopía óptica con muy alta resolución que puede obtener imágenes de detalles en muestras que no se pueden obtener con microscopía convencional o confocal . Dentro de RESOLFT se generalizan los principios de microscopía STED y microscopía GSD . Además, existen técnicas con conceptos distintos a RESOLFT o SSIM. Por ejemplo, microscopía de fluorescencia que utiliza la propiedad de puerta óptica AND del centro de vacancia de nitrógeno , o superresolución por emisión estimulada de radiación térmica (SETR), que utiliza las superlinearidades intrínsecas de la radiación de cuerpo negro y amplía el concepto de superresolución. -resolución más allá de la microscopía.
Agotamiento de emisiones estimulado (STED)

La microscopía de agotamiento de emisión estimulada (STED) utiliza dos pulsos láser, el pulso de excitación para la excitación de los fluoróforos a su estado fluorescente y el pulso STED para la desexcitación de los fluoróforos mediante emisión estimulada . En la práctica, el pulso de láser de excitación se aplica primero, después de lo cual sigue pronto un pulso STED (también se usa STED sin pulsos que utilizan láseres de onda continua). Además, el pulso STED se modifica de tal manera que presenta un punto de intensidad cero que coincide con el punto focal de excitación. Debido a la dependencia no lineal de la tasa de emisión estimulada de la intensidad del haz STED, todos los fluoróforos alrededor del punto de excitación focal estarán en su estado desactivado (el estado fundamental de los fluoróforos). Al escanear este punto focal, se recupera la imagen. El ancho completo a la mitad del máximo (FWHM) de la función de dispersión de puntos (PSF) del punto focal de excitación se puede comprimir teóricamente a un ancho arbitrario aumentando la intensidad del pulso STED, de acuerdo con la ecuación ( 1 ).
- ( 1 )

- donde ∆r es la resolución lateral, ∆ es la FWHM de la PSF limitada por difracción, I max es la intensidad máxima del láser STED y es la intensidad umbral necesaria para lograr el agotamiento de las emisiones saturadas.

La principal desventaja de STED, que ha impedido su uso generalizado, es que la maquinaria es complicada. Por un lado, la velocidad de adquisición de imágenes es relativamente lenta para campos de visión grandes debido a la necesidad de escanear la muestra para recuperar una imagen. Por otro lado, puede ser muy rápido para campos de visión más pequeños: se han mostrado grabaciones de hasta 80 fotogramas por segundo. Debido a la gran I s valor asociado con STED, existe la necesidad de un impulso de excitación de alta intensidad, que puede causar daño a la muestra.
Agotamiento del estado fundamental (GSD)
La microscopía de agotamiento del estado fundamental ( microscopía GSD) utiliza el estado triplete de un fluoróforo como estado desactivado y el estado singlete como estado activado, por lo que se utiliza un láser de excitación para impulsar los fluoróforos en la periferia de la molécula en estado singlete a la estado triplete. Esto es muy parecido a STED, donde el estado desactivado es el estado fundamental de los fluoróforos, por lo que la ecuación ( 1 ) también se aplica en este caso. El valor es menor que en STED, lo que permite obtener imágenes de superresolución con una intensidad de láser mucho menor. Sin embargo, en comparación con STED, los fluoróforos utilizados en GSD son generalmente menos fotoestables; y la saturación del estado triplete puede ser más difícil de realizar.
Microscopía de iluminación estructurada saturada (SSIM)
La microscopía de iluminación estructurada saturada (SSIM) aprovecha la dependencia no lineal de la tasa de emisión de fluoróforos de la intensidad del láser de excitación. Al aplicar un patrón de iluminación sinusoidal con una intensidad máxima cercana a la necesaria para saturar los fluoróforos en su estado fluorescente, se recuperan las franjas de Moiré. Las franjas contienen información espacial de alto orden que puede extraerse mediante técnicas computacionales. Una vez que se extrae la información, se recupera una imagen de superresolución.
SSIM requiere cambiar el patrón de iluminación varias veces, limitando efectivamente la resolución temporal de la técnica. Además, existe la necesidad de fluoróforos muy fotoestables, debido a las condiciones de saturación, que infligen daños por radiación en la muestra y restringen las posibles aplicaciones para las que se puede utilizar SSIM.
Se muestran ejemplos de esta microscopía en la sección Microscopía de iluminación estructurada (SIM) : imágenes de núcleos celulares y etapas mitóticas registradas con microscopía 3D-SIM.
Técnicas funcionales estocásticas
Microscopía de localización
La microscopía de localización de una sola molécula (SMLM) resume todas las técnicas microscópicas que logran una superresolución al aislar los emisores y ajustar sus imágenes con la función de dispersión de puntos (PSF). Normalmente, el ancho de la función de dispersión de puntos (~ 250 nm) limita la resolución. Sin embargo, dado un emisor aislado, se puede determinar su ubicación con una precisión solo limitada por su intensidad de acuerdo con la ecuación ( 2 ).
- ( 2 )

- donde Δloc es la precisión de localización, Δ es el FWHM (ancho completo a la mitad del máximo) de la PSF y N es el número de fotones recolectados.
Este proceso de ajuste solo se puede realizar de manera confiable para emisores aislados (ver Deconvolución ), y las muestras biológicas interesantes están tan densamente etiquetadas con emisores que el ajuste es imposible cuando todos los emisores están activos al mismo tiempo. Las técnicas SMLM resuelven este dilema activando solo un escaso subconjunto de emisores al mismo tiempo, localizando estos pocos emisores con mucha precisión, desactivándolos y activando otro subconjunto.
Considerando el fondo y la pixelación de la cámara, y usando la aproximación gaussiana para la función de dispersión de puntos ( disco de Airy ) de un microscopio típico, la resolución teórica es propuesta por Thompson et al. y perfeccionado por Mortensen et al .:

- dónde
- * σ es la desviación estándar gaussiana de las ubicaciones centrales de la misma molécula si se mide varias veces (por ejemplo, fotogramas de un video). (unidad m)
- * σ PSF es la desviación estándar gaussiana de la función de dispersión de puntos, cuyo FWHM siguiendo la ecuación de Ernst Abbe d = λ / (2 NA) . (unidad m)
- * a es el tamaño de cada píxel de la imagen. (unidad m)
- * N sig son los recuentos de fotones del PSF total en todos los píxeles de interés. (sin unidad)
- * N bg el recuento medio de fotones de fondo por píxel (recuentos oscuros ya eliminados), que se aproxima al cuadrado de la desviación estándar gaussiana del ruido de fondo de la distribución de Poisson de cada píxel a lo largo del tiempo o la desviación estándar de todos los píxeles con solo ruido de fondo , σ bg 2 . Cuanto mayor sea σ bg 2 , mejor será la aproximación (por ejemplo, buena para σ bg 2 > 10, excelente para σ bg 2 > 1000). (sin unidad)
- * La resolución FWHM es ~ 2,355 veces la desviación estándar de Gauss .
Generalmente, la microscopía de localización se realiza con fluoróforos. Los fluoróforos adecuados (por ejemplo, para STORM) residen en un estado oscuro no fluorescente durante la mayor parte del tiempo y se activan estocásticamente, típicamente con un láser de excitación de baja intensidad. Un láser de lectura estimula la fluorescencia y blanquea o fotoconmuta los fluoróforos de nuevo a un estado oscuro, por lo general en 10 a 100 ms. En la acumulación de puntos para la obtención de imágenes en topografía a nanoescala (PAINT), los fluoróforos no son fluorescentes antes de unirse y son fluorescentes después. Los fotones emitidos durante la fase fluorescente se recogen con una cámara y la imagen resultante del fluoróforo (que está distorsionada por la PSF) se puede ajustar con una precisión muy alta, incluso del orden de unos pocos Angstroms. Repetir el proceso varios miles de veces asegura que todos los fluoróforos puedan pasar por el estado brillante y se registren. Luego, una computadora reconstruye una imagen súper resuelta.
Los rasgos deseables de los fluoróforos utilizados para estos métodos, con el fin de maximizar la resolución, son que deben ser brillantes. Es decir, deberían tener un alto coeficiente de extinción y un alto rendimiento cuántico . También deben poseer una alta relación de contraste (relación entre el número de fotones emitidos en el estado de luz y el número de fotones emitidos en el estado de oscuridad). Además, es deseable una muestra densamente etiquetada, de acuerdo con los criterios de Nyquist .
La multitud de métodos de microscopía de localización difieren principalmente en el tipo de fluoróforos utilizados.
Microscopía de distancia de precisión espectral (SPDM)

Una única y diminuta fuente de luz se puede ubicar mucho mejor de lo que la resolución de un microscopio suele permitir: aunque la luz producirá un punto borroso, se pueden usar algoritmos informáticos para calcular con precisión el centro del punto borroso, teniendo en cuenta la la función de dispersión puntual del microscopio, las propiedades de ruido del detector, etc. Sin embargo, este enfoque no funciona cuando hay demasiadas fuentes cercanas entre sí: las fuentes se difuminan todas juntas.
La microscopía de distancia de precisión espectral (SPDM) es una familia de técnicas de localización en microscopía de fluorescencia que resuelve el problema de que hay muchas fuentes midiendo solo unas pocas fuentes a la vez, de modo que cada fuente está "ópticamente aislada" de las demás (es decir, , separados por más de la resolución del microscopio, típicamente ~ 200-250 nm), si las partículas bajo examen tienen diferentes firmas espectrales, de modo que sea posible observar la luz de unas pocas moléculas a la vez utilizando las fuentes de luz adecuadas. y filtros. Esto logra una resolución óptica efectiva varias veces mejor que la resolución óptica convencional que está representada por la mitad del ancho del máximo principal de la función de imagen puntual efectiva.
La resolución estructural que se puede lograr usando SPDM se puede expresar en términos de la menor distancia medible entre dos partículas puntiformes de diferentes características espectrales ("resolución topológica"). El modelado ha demostrado que en condiciones adecuadas en cuanto a precisión de localización, densidad de partículas, etc., la "resolución topológica" corresponde a una " frecuencia espacial " que, en términos de la definición clásica, equivale a una resolución óptica muy mejorada. Las moléculas también se pueden distinguir de formas aún más sutiles basadas en la vida útil fluorescente y otras técnicas.
Una aplicación importante es la investigación del genoma (estudio de la organización funcional del genoma ). Otro campo de aplicación importante es la investigación de la estructura de las membranas.
SPDMphymod

La microscopía de localización para muchos tintes fluorescentes estándar como GFP , tintes Alexa y moléculas de fluoresceína es posible si están presentes ciertas condiciones fotofísicas. Con esta tecnología denominada fluoróforos físicamente modificables (SPDMphymod) , una sola longitud de onda láser de intensidad adecuada es suficiente para la obtención de nano imágenes en contraste con otras tecnologías de microscopía de localización que necesitan dos longitudes de onda láser cuando se utilizan moléculas especiales de fluorescencia fotoconmutables / fotoactivables. Otro ejemplo del uso de SPDMphymod es un análisis de partículas del virus del mosaico del tabaco (TMV) o el estudio de la interacción virus-célula .
Sobre la base de las transiciones de estado singlete-triplete, es crucial para SPDMphymod que este proceso sea continuo y que lleve al efecto de que una sola molécula entre primero en un estado oscuro reversible de larga duración (con una vida media de hasta varios segundos) desde que vuelve a un estado fluorescente que emite muchos fotones durante varios milisegundos antes de volver a un estado oscuro muy prolongado, llamado irreversible. La microscopía SPDMphymod utiliza moléculas fluorescentes que emiten la misma frecuencia de luz espectral pero con diferentes firmas espectrales según las características de destello. Al combinar dos mil imágenes de la misma celda, es posible, utilizando mediciones de precisión óptica láser, registrar imágenes de localización con una resolución óptica significativamente mejorada.
Los tintes fluorescentes estándar que ya se han utilizado con éxito con la tecnología SPDMphymod son GFP , RFP , YFP , Alexa 488 , Alexa 568, Alexa 647, Cy2 , Cy3, Atto 488 y fluoresceína .
Localización óptica criogénica en 3D (COLD)

La localización óptica criogénica en 3D (COLD) es un método que permite localizar múltiples sitios fluorescentes dentro de una única biomolécula de tamaño pequeño a mediano con resolución de escala Angstrom. La precisión de localización en este enfoque se mejora porque la fotoquímica más lenta a bajas temperaturas conduce a un mayor número de fotones que se pueden emitir desde cada fluoróforo antes del fotoblanqueo. Como resultado, la microscopía de localización estocástica criogénica logra la resolución submolecular requerida para resolver las posiciones 3D de varios fluoróforos unidos a una proteína pequeña. Empleando algoritmos conocidos de microscopía electrónica, las proyecciones 2D de fluoróforos se reconstruyen en una configuración 3D. COLD lleva la microscopía de fluorescencia a su límite fundamental, dependiendo del tamaño de la etiqueta. El método también se puede combinar con otras técnicas de biología estructural, como la cristalografía de rayos X, la espectroscopia de resonancia magnética y la microscopía electrónica, para proporcionar una valiosa información complementaria y especificidad.
Microscopía de localización activada por unión (BALM)

La microscopía de localización activada por unión (BALM) es un concepto general para la microscopía de localización de una sola molécula (SMLM): obtención de imágenes súper resueltas de colorantes de unión al ADN basadas en la modificación de las propiedades del ADN y un colorante. Mediante un ajuste cuidadoso del entorno químico, lo que conduce a un control de hibridación y fusión del ADN reversible y local sobre la señal de fluorescencia, se pueden introducir moléculas de colorante que se unen al ADN. Pueden usarse colorantes de ADN de unión intercalada y de surco menor para registrar y aislar ópticamente solo unas pocas señales de colorante de unión a ADN a la vez. El BALM asistido por fluctuación de la estructura del ADN (fBALM) se ha utilizado para diferencias a nanoescala en la arquitectura nuclear, con una resolución estructural anticipada de aproximadamente 50 nm. Imágenes de nanoestructura de cromatina con microscopía de localización activada por unión basada en fluctuaciones de la estructura del ADN. Recientemente, se aprovechó la mejora significativa del rendimiento cuántico de fluorescencia de NIAD-4 tras la unión a un amiloide para la formación de imágenes BALM de fibrillas y oligómeros amiloides.
TORMENTA, PALMA y FPALM
La microscopía de reconstrucción óptica estocástica (STORM), la microscopía de localización fotoactivada (PALM) y la microscopía de localización por fotoactivación de fluorescencia (FPALM) son técnicas de imágenes de superresolución que utilizan la activación secuencial y la localización resuelta en el tiempo de fluoróforos fotoconmutables para crear imágenes de alta resolución. Durante la formación de imágenes, solo un subconjunto de fluoróforos que se puede resolver ópticamente se activa a un estado fluorescente en un momento dado, de modo que la posición de cada fluoróforo se puede determinar con alta precisión al encontrar las posiciones del centroide de las imágenes de una sola molécula de un fluoróforo en particular. Posteriormente, se desactiva un subconjunto de fluoróforos y se activa y se obtienen imágenes de otro subconjunto. La iteración de este proceso permite localizar numerosos fluoróforos y construir una imagen de superresolución a partir de los datos de la imagen.
Estos tres métodos se publicaron de forma independiente durante un breve período de tiempo y sus principios son idénticos. STORM se describió originalmente usando tintes Cy5 y Cy3 unidos a ácidos nucleicos o proteínas, mientras que PALM y FPALM se describieron usando proteínas fluorescentes fotoconmutables. En principio, se puede usar cualquier fluoróforo fotoconmutable, y STORM se ha demostrado con una variedad de diferentes sondas y estrategias de marcado. Usando la conmutación fotográfica estocástica de fluoróforos individuales, como Cy5, se puede realizar STORM con una sola fuente de excitación de láser rojo. El láser rojo cambia el fluoróforo Cy5 a un estado oscuro mediante la formación de un aducto y posteriormente devuelve la molécula al estado fluorescente. También se han utilizado muchos otros tintes con STORM.
Además de los fluoróforos individuales, los pares de tintes que consisten en un fluoróforo activador (como Alexa 405, Cy2 o Cy3) y un tinte reportero fotoconmutable (como Cy5, Alexa 647, Cy5.5 o Cy7) se pueden usar con STORM . En este esquema, el fluoróforo activador, cuando se excita cerca de su máximo de absorción, sirve para reactivar el tinte fotoconmutable al estado fluorescente. Se han realizado imágenes multicolores usando diferentes longitudes de onda de activación para distinguir pares de tintes, dependiendo del fluoróforo activador usado, o usando fluoróforos fotoconmutables espectralmente distintos, con o sin fluoróforos activadores. También se pueden usar proteínas fluorescentes fotoconmutables. Se ha logrado un marcaje altamente específico de estructuras biológicas con sondas fotoconmutables con tinción de anticuerpos, conjugación directa de proteínas y codificación genética.
STORM también se ha extendido a imágenes tridimensionales utilizando astigmatismo óptico, en el que la forma elíptica de la función de dispersión de puntos codifica las posiciones x, y, z para muestras de hasta varios micrómetros de espesor, y se ha demostrado en células vivas. Hasta la fecha, la resolución espacial lograda por esta técnica es ~ 20 nm en las dimensiones laterales y ~ 50 nm en la dimensión axial; y la resolución temporal es tan rápida como 0,1-0,33 s.
Acumulación de puntos para la obtención de imágenes en topografía a nanoescala (PAINT)
La acumulación de puntos para la obtención de imágenes en topografía a nanoescala (PAINT) es un método de localización de una sola molécula que logra una fluorescencia estocástica de una sola molécula mediante adsorción / absorción molecular y fotoblanqueo / desorción. El primer tinte utilizado fue el rojo del Nilo, que no es fluorescente en solución acuosa, pero sí fluorescente cuando se inserta en un entorno hidrófobo, como micelas o paredes de células vivas. Por lo tanto, la concentración del tinte se mantiene pequeña, en el nivel nanomolar, de modo que la tasa de sorción de la molécula al área limitada por difracción está en la región de milisegundos. La unión estocástica de moléculas de un solo colorante (sondas) a un objetivo inmovilizado puede resolverse espacial y temporalmente bajo un microscopio de fluorescencia de campo amplio típico. Cada tinte se fotoblanquea para devolver el campo a un estado oscuro, de modo que el siguiente tinte pueda unirse y ser observado. La ventaja de este método, en comparación con otros métodos estocásticos, es que además de obtener la imagen superesuelta del objetivo fijo, puede medir la cinética de unión dinámica de las moléculas de la sonda de difusión, en solución, al objetivo.
Combinando la técnica de superresolución 3D (por ejemplo, la función de dispersión de puntos de doble hélice desarrollada en el grupo de Moerner), tintes fotoactivados, intermitencia activa dependiente de la potencia y acumulación de puntos para obtener imágenes en topografía a nanoescala, SPRAIPAINT (SPRAI = Super resolución de PoweR- intermitencia activa dependiente) puede superresolver las paredes de células vivas. PAINT funciona manteniendo un equilibrio entre las tasas de adsorción / absorción del tinte y de fotoblanqueo / desorción. Este saldo se puede estimar con principios estadísticos. La tasa de adsorción o absorción de un soluto diluido a una superficie o interfaz en una solución gaseosa o líquida se puede calcular utilizando las leyes de difusión de Fick . La tasa de fotoblanqueo / desorción se puede medir para una determinada condición de solución y densidad de potencia de iluminación.
DNA-PAINT se ha ampliado aún más para utilizar tintes regulares, donde se utiliza la unión y desvinculación dinámica de una sonda de ADN marcada con un tinte a un origami de ADN fijo para lograr imágenes estocásticas de una sola molécula. DNA-PAINT ya no se limita a los tintes sensibles al medio ambiente y puede medir la cinética de adsorción y desorción de las sondas al objetivo. El método utiliza el efecto de desenfoque de la cámara de los tintes en movimiento. Cuando un tinte normal se difunde en la solución, su imagen en una cámara CCD típica se ve borrosa debido a su velocidad relativamente rápida y al tiempo de exposición de la cámara relativamente largo, lo que contribuye al fondo de fluorescencia. Sin embargo, cuando se une a un objetivo fijo, el tinte deja de moverse; y se puede lograr una entrada clara en la función de dispersión de puntos.
El término para este método es mbPAINT ("mb" significa desenfoque de movimiento ). Cuando se utiliza un microscopio de fluorescencia de reflexión interna total (TIRF) para obtener imágenes, la profundidad de excitación se limita a ~ 100 nm desde el sustrato, lo que reduce aún más el fondo de fluorescencia de los tintes borrosos cerca del sustrato y el fondo en la solución a granel. Se pueden usar tintes muy brillantes para mbPAINT, que proporciona resoluciones espaciales típicas de un solo cuadro de ~ 20 nm y resoluciones temporales cinéticas de una sola molécula de ~ 20 ms bajo intensidades de fotoexcitación relativamente suaves, lo que es útil para estudiar la separación molecular de proteínas individuales.
La resolución temporal se ha mejorado aún más (20 veces) utilizando una máscara de fase rotacional colocada en el plano de Fourier durante la adquisición de datos y resolviendo la función de dispersión de puntos distorsionados que contiene información temporal. El método se denominó Microscopía Súper Temporal-Resuelta (STReM).
Microscopía de localización sin etiquetas

Se puede lograr una resolución óptica de estructuras celulares en el rango de aproximadamente 50 nm, incluso en células sin marcaje, usando microscopía de localización SPDM .
Mediante el uso de dos longitudes de onda láser diferentes, SPDM revela objetos celulares que no son detectables en condiciones de imágenes de campo amplio de fluorescencia convencionales, además de lograr una mejora sustancial de la resolución de las estructuras autofluorescentes.
Como control, las posiciones de los objetos detectados en la imagen de localización coinciden con las de la imagen de campo claro.
La microscopía de superresolución sin etiquetas también se ha demostrado utilizando las fluctuaciones de una señal de dispersión Raman mejorada en la superficie en una metasuperficie plasmónica altamente uniforme.
Microscopía de reconstrucción óptica estocástica directa (dSTORM)
dSTORM utiliza la fotoconmutación de un solo fluoróforo. En dSTORM, los fluoróforos se incrustan en un sistema tampón reductor y oxidante (ROXS) y se excita la fluorescencia. A veces, estocásticamente, el fluoróforo entrará en un triplete o en algún otro estado oscuro que sea sensible al estado de oxidación del tampón, a partir del cual pueden hacerse fluorescentes, de modo que se puedan registrar las posiciones de las moléculas individuales. El desarrollo del método dSTORM se produjo en 3 laboratorios independientes aproximadamente al mismo tiempo y también se denominó "microscopía de fotoblanqueo reversible" (RPM), "microscopía de agotamiento del estado fundamental seguida de retorno de moléculas individuales" (GSDIM), así como la ahora generalmente aceptada apodo dSTORM.
Software para microscopía de localización
La microscopía de localización depende en gran medida de un software que pueda ajustar con precisión la función de dispersión puntual (PSF) a millones de imágenes de fluoróforos activos en unos pocos minutos. Dado que los métodos de análisis clásicos y los paquetes de software utilizados en las ciencias naturales son demasiado lentos para resolver estos problemas computacionalmente, y a menudo se requieren horas de cálculo para procesar datos medidos en minutos, se han desarrollado programas de software especializados. Muchos de estos paquetes de software de localización son de código abierto; se enumeran en SMLM Software Benchmark. Una vez que se han determinado las posiciones de las moléculas, es necesario mostrar las ubicaciones y se han desarrollado varios algoritmos para la visualización.
Imágenes de fluctuación óptica de súper resolución (SOFI)
Es posible eludir la necesidad de ajuste de PSF inherente a la microscopía de localización de molécula única (SMLM) calculando directamente la autocorrelación temporal de píxeles. Esta técnica se denomina imagen de fluctuación óptica de superresolución (SOFI) y se ha demostrado que es más precisa que SMLM cuando la densidad de fluoróforos activos al mismo tiempo es muy alta.
Microscopía de localización omnipresente (OLM)
La microscopía de localización omnipresente (OLM) es una extensión de las técnicas de microscopía de molécula única (SMLM) que permiten obtener imágenes de una sola molécula de alta densidad con una fuente de luz incoherente (como una lámpara de arco de mercurio) y una configuración de microscopio de epifluorescencia convencional. Una breve ráfaga de excitación azul profundo (con un láser de 350-380 nm, en lugar de 405 nm) permite una reactivación prolongada de moléculas, para una resolución de 90 nm en las muestras de prueba. Finalmente, las imágenes correlativas de STED y SMLM se pueden realizar en la misma muestra biológica utilizando un medio de imagen simple, que puede proporcionar una base para una resolución mejorada adicional. Estos hallazgos pueden democratizar las imágenes de superresolución y ayudar a cualquier científico a generar imágenes de una sola molécula de alta densidad incluso con un presupuesto limitado.
Combinación de técnicas
Microscopía de nanoescala microscópica de luz 3D (LIMON)

Las imágenes de microscopía de nanoescala microscópica de luz (3D LIMON), utilizando el microscopio Vertico SMI , son posibles gracias a la combinación de SMI y SPDM , mediante el cual primero se aplica el proceso SMI y luego el SPDM.
El proceso SMI determina el centro de las partículas y su propagación en la dirección del eje del microscopio. Si bien el centro de partículas / moléculas se puede determinar con una precisión de 1 a 2 nm, la dispersión alrededor de este punto se puede determinar hasta un diámetro axial de aproximadamente 30 a 40 nm.
Posteriormente, se determina la posición lateral de la partícula / molécula individual mediante SPDM, logrando una precisión de unos pocos nanómetros.
Como una aplicación biológica en el modo de color dual 3D, se logró la disposición espacial de los grupos Her2 / neu y Her3 . Las posiciones en las tres direcciones de los grupos de proteínas se pudieron determinar con una precisión de aproximadamente 25 nm.
Microscopía electrónica y de luz correlativa integrada
La combinación de un microscopio de superresolución con un microscopio electrónico permite la visualización de información contextual, con el etiquetado proporcionado por marcadores de fluorescencia. Esto supera el problema del fondo negro con el que se queda el investigador cuando usa solo un microscopio óptico. En un sistema integrado, la muestra se mide con ambos microscopios simultáneamente.
Mejora de técnicas mediante redes neuronales
Recientemente, debido a los avances en la computación de inteligencia artificial, las redes neuronales de aprendizaje profundo se han utilizado para mejorar la superresolución de imágenes fotográficas de microscopio óptico, de 40x a 100x, de 20x con un microscopio óptico a 1500x, comparable a un microscopio electrónico de barrido. , a través de una lente neural, y con tomografía por emisión de positrones y microscopía de fluorescencia.
Ver también
- Microscopía de plano multifocal (MUM)
- Microscopio de reducción de emisiones estimuladas (STED)
- Microscopía de localización fotoactivada (PALM)
- Microscopía de reconstrucción óptica estocástica (STORM)
- Deconvolución
- Sondas fotoactivables
- Microscopía electrónica de luz correlativa
- Imágenes de súper resolución
- Super resolución de video
Referencias
Otras lecturas
- Marx V (diciembre de 2013) [26 de noviembre de 2013]. "¿Es la microscopía de superresolución adecuada para usted?". Característica tecnológica. Métodos de la naturaleza (artículo "Colección de reimpresiones de la naturaleza, características tecnológicas"). 10 (12): 1157–63. doi : 10.1038 / nmeth.2756 . PMID 24296472 . S2CID 1004998 .
- Cremer C, Masters BR (abril de 2013). "Técnicas de mejora de resolución en microscopía" . El European Physical Diario H . 38 (3): 281–344. Código Bibliográfico : 2013EPJH ... 38..281C . doi : 10.1140 / epjh / e2012-20060-1 .