
Púrpura trombocitopénica inmunitaria - Immune thrombocytopenic purpura
| Púrpura trombocitopénica inmunitaria | |
|---|---|
| Otros nombres | Púrpura trombocitopénica idiopática, trombocitopenia inmune idiopática, trombocitopenia inmune primaria, púrpura trombocitopénica idiopática, púrpura trombocitopénica inmune primaria, púrpura trombocitopénica autoinmune |
 | |
| En la PTI pueden aparecer petequias o pequeñas marcas en forma de hematoma. | |
| Especialidad | Hematología , cirugía general |
La púrpura trombocitopénica inmunitaria ( PTI ), también conocida como púrpura trombocitopénica idiopática o trombocitopenia inmunitaria , es un tipo de púrpura trombocitopénica definida como un recuento bajo de plaquetas aislado con médula ósea normal en ausencia de otras causas de plaquetas bajas. Provoca una erupción característica de color rojo o morado similar a un hematoma y una mayor tendencia a sangrar. Dos síndromes clínicos distintos se manifiestan como una afección aguda en los niños y una afección crónica en los adultos. La forma aguda a menudo sigue a una infección y se resuelve espontáneamente en dos meses. La trombocitopenia inmunitaria crónica persiste durante más de seis meses y se desconoce una causa específica.
La PTI es una enfermedad autoinmune con anticuerpos detectables contra varias estructuras de la superficie plaquetaria .
La PTI se diagnostica identificando un recuento bajo de plaquetas en un hemograma completo (un análisis de sangre común ). Sin embargo, dado que el diagnóstico depende de la exclusión de otras causas de un recuento bajo de plaquetas, en algunos casos pueden ser necesarias investigaciones adicionales (como una biopsia de médula ósea ).
En los casos leves, es posible que solo se requiera una observación cuidadosa, pero los recuentos muy bajos o el sangrado significativo pueden impulsar el tratamiento con corticosteroides , inmunoglobulina intravenosa , inmunoglobulina anti-D o medicamentos inmunosupresores . La PTI refractaria (que no responde al tratamiento convencional o recaída constante después de la esplenectomía ) requiere tratamiento para reducir el riesgo de hemorragia clínicamente significativa. Las transfusiones de plaquetas se pueden usar en casos graves con recuentos de plaquetas muy bajos en personas que están sangrando. A veces, el cuerpo puede compensarlo produciendo plaquetas anormalmente grandes.
Signos y síntomas
Los signos incluyen la formación espontánea de hematomas (púrpura) y petequias (pequeños hematomas), especialmente en las extremidades , sangrado de las fosas nasales y / o encías , y menorragia ( sangrado menstrual excesivo ), cualquiera de los cuales puede ocurrir si el recuento de plaquetas es inferior. 20.000 por μl. Un recuento muy bajo (<10.000 por μl) puede provocar la formación espontánea de hematomas (masas de sangre) en la boca o en otras membranas mucosas . El tiempo de sangrado por laceraciones o abrasiones menores suele ser prolongado.
Las complicaciones graves y posiblemente fatales debido a recuentos extremadamente bajos (<5,000 por μl) incluyen hemorragia subaracnoidea o intracerebral (hemorragia dentro del cráneo o cerebro ), hemorragia digestiva baja u otra hemorragia interna. Un paciente con PTI con un recuento extremadamente bajo es vulnerable a una hemorragia interna causada por un traumatismo abdominal cerrado , como se podría experimentar en un accidente automovilístico . Estas complicaciones no son probables cuando el recuento de plaquetas es superior a 20.000 por μl.
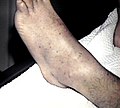
Petequias en las extremidades inferiores.
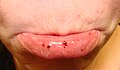
Petequias orales / púrpura - labio inferior
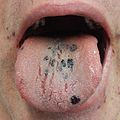
Petequias en la lengua en una persona con 3 plaquetas por PTI

Petequias de la parte inferior de la pierna en una persona con 3 plaquetas por PTI




Patogénesis
En aproximadamente el 60 por ciento de los casos, se pueden detectar anticuerpos contra las plaquetas. Muy a menudo, estos anticuerpos son contra las glicoproteínas de la membrana plaquetaria IIb-IIIa o Ib-IX , y son del tipo inmunoglobulina G (IgG). El experimento de Harrington-Hollingsworth estableció la patogenia inmunológica de la PTI.
El recubrimiento de las plaquetas con IgG las hace susceptibles a la opsonización y fagocitosis por los macrófagos esplénicos , así como por las células de Kupffer en el hígado . También se cree que los autoanticuerpos IgG dañan los megacariocitos , las células precursoras de las plaquetas, aunque se cree que esto contribuye sólo ligeramente a la disminución del número de plaquetas. Investigaciones recientes indican ahora que la producción deficiente de la hormona glicoproteica trombopoyetina , que es el estimulante de la producción de plaquetas, puede ser un factor que contribuya a la reducción de las plaquetas circulantes. Esta observación ha llevado al desarrollo de una clase de medicamentos dirigidos a la PTI denominados agonistas del receptor de trombopoyetina .
El estímulo para la producción de autoanticuerpos en la PTI es probablemente la actividad anormal de las células T. Los hallazgos preliminares sugieren que estas células T pueden verse influenciadas por medicamentos que se dirigen a las células B , como el rituximab .
Diagnóstico

El diagnóstico de PTI es un proceso de exclusión. En primer lugar, se debe determinar que no hay anomalías en la sangre que no sean un recuento bajo de plaquetas, y que no hay signos físicos aparte de sangrado. Luego, deben excluirse las causas secundarias (5 a 10 por ciento de los casos sospechosos de PTI). Tales causas secundarias incluyen leucemia , medicamentos (p. Ej., Quinina , heparina ), lupus eritematoso , cirrosis , VIH , hepatitis C, causas congénitas, síndrome antifosfolípido , deficiencia del factor von Willebrand , onyalai y otras. Todos los pacientes con presunta PTI deben someterse a pruebas de detección del VIH y el virus de la hepatitis C, ya que el recuento de plaquetas puede corregirse mediante el tratamiento de la enfermedad subyacente. En aproximadamente el 2,7 al 5 por ciento de los casos, coexisten anemia hemolítica autoinmune e ITP, una condición conocida como síndrome de Evans .
A pesar de la destrucción de las plaquetas por los macrófagos esplénicos, el bazo normalmente no está agrandado. De hecho, un bazo agrandado debería conducir a la búsqueda de otras posibles causas de la trombocitopenia. El tiempo de sangrado suele prolongarse en pacientes con PTI. Sin embargo, las guías de práctica de la Sociedad Estadounidense de Hematología desaconsejan el uso del tiempo de sangrado en el diagnóstico y un tiempo de sangrado normal no excluye un trastorno plaquetario.
El examen de la médula ósea se puede realizar en pacientes mayores de 60 años y aquellos que no responden al tratamiento, o cuando el diagnóstico es dudoso. En el examen de la médula, se puede observar un aumento en la producción de megacariocitos y puede ayudar a establecer un diagnóstico de PTI. Un análisis de anticuerpos antiplaquetarios es una cuestión de preferencia del médico, ya que existe un desacuerdo sobre si el 80 por ciento de especificidad de esta prueba es suficiente para ser clínicamente útil.
Tratamiento
Con raras excepciones, por lo general no es necesario un tratamiento basado en el recuento de plaquetas. Muchas recomendaciones anteriores sugirieron un cierto umbral de recuento de plaquetas (generalmente en algún lugar por debajo de 20,0 / µl) como indicación de hospitalización o tratamiento. Las pautas actuales recomiendan el tratamiento solo en casos de hemorragia significativa. Las recomendaciones de tratamiento a veces difieren para la PTI en adultos y en niños.
Esteroides
El tratamiento inicial generalmente consiste en la administración de corticosteroides , un grupo de medicamentos que inhiben el sistema inmunológico. La dosis y el modo de administración se determinan por el recuento de plaquetas y si hay sangrado activo: en situaciones urgentes, se pueden usar infusiones de dexametasona o metilprednisolona , mientras que prednisona o prednisolona orales pueden ser suficientes en casos menos graves. Una vez que ha mejorado el recuento de plaquetas, la dosis de esteroide se reduce gradualmente mientras se controla la posibilidad de recaída. El 60-90 por ciento experimentará una recaída durante la reducción o el cese de la dosis. Los esteroides a largo plazo se evitan si es posible debido a los posibles efectos secundarios que incluyen osteoporosis , diabetes y cataratas .
Anti-D
Otra opción, adecuada para pacientes Rh positivos con bazos funcionales es la administración intravenosa de inmunoglobulina Rho (D) [Human; Anti-D]. El mecanismo de acción de anti-D no se comprende completamente. Sin embargo, después de la administración, los complejos de eritrocitos recubiertos con anti-D saturan los sitios del receptor de Fcγ en los macrófagos , lo que da como resultado una destrucción preferencial de los glóbulos rojos (RBC) y, por lo tanto, evitan las plaquetas recubiertas de anticuerpos . Hay dos productos anti-D indicados para su uso en pacientes con PTI: WinRho SDF y Rhophylac. Las reacciones adversas más frecuentes son dolor de cabeza (15%), náuseas / vómitos (12%) escalofríos (<2%) y fiebre (1%).
Agentes ahorradores de esteroides
Existe un uso creciente de inmunosupresores como el micofenolato de mofetilo y la azatioprina debido a su eficacia. En casos refractarios crónicos, en los que se ha confirmado la patogenia inmunitaria, se puede intentar el uso no autorizado del alcaloide de la vinca y el agente quimioterapéutico vincristina . Sin embargo, la vincristina tiene efectos secundarios importantes y su uso en el tratamiento de la PTI debe abordarse con precaución, especialmente en niños.
Inmunoglobulina intravenosa
En algunos casos, se puede infundir inmunoglobulina intravenosa (IgIV) para disminuir la velocidad a la que los macrófagos consumen plaquetas marcadas con anticuerpos . Sin embargo, aunque a veces es efectivo, es costoso y produce mejoras que generalmente duran menos de un mes. Sin embargo, en el caso de un paciente con PTI ya programado para una cirugía que tiene un recuento de plaquetas peligrosamente bajo y ha experimentado una mala respuesta a otros tratamientos, la IgIV puede aumentar rápidamente el recuento de plaquetas y también puede ayudar a reducir el riesgo de hemorragia mayor al aumentar transitoriamente recuentos de plaquetas.
Agonistas del receptor de trombopoyetina
Los agonistas del receptor de trombopoyetina son agentes farmacéuticos que estimulan la producción de plaquetas en la médula ósea. En esto, difieren de los agentes discutidos anteriormente que actúan intentando reducir la destrucción de plaquetas. Actualmente hay dos productos de este tipo disponibles:
- Romiplostim (nombre comercial Nplate) es una proteína de fusión Fc-péptido estimulante de la trombopoyesis (pepticuerpo) que se administra por inyección subcutánea . Designado como medicamento huérfano en 2003 según la ley de los Estados Unidos, los ensayos clínicos demostraron que el romiplostim es eficaz en el tratamiento de la PTI crónica, especialmente en pacientes con recaída después de la esplenectomía. El romiplostim fue aprobado por la Administración de Drogas y Alimentos de los Estados Unidos (FDA) para el tratamiento a largo plazo de la PTI crónica en adultos el 22 de agosto de 2008.
- Eltrombopag (nombre comercial Promacta en EE. UU., Revolade en la UE) es un fármaco administrado por vía oral con un efecto similar al del romiplostim. También se ha demostrado que aumenta el recuento de plaquetas y reduce el sangrado de manera dependiente de la dosis. Desarrollado por GlaxoSmithKline y también designado como medicamento huérfano por la FDA, Promacta fue aprobado por la FDA el 20 de noviembre de 2008.
Los agonistas del receptor de trombopoyetina exhibieron el mayor éxito hasta ahora en el tratamiento de pacientes con PTI refractaria.
Los efectos secundarios de los agonistas del receptor de trombopoyetina incluyen dolor de cabeza, dolor articular o muscular, mareos, náuseas o vómitos y un mayor riesgo de coágulos sanguíneos.
Cirugía
Se puede considerar la esplenectomía (extirpación del bazo ) en pacientes que no responden al tratamiento con esteroides, que tienen recaídas frecuentes o que no pueden dejar de tomar esteroides después de unos meses. Las plaquetas unidas por anticuerpos son captadas por macrófagos en el bazo (que tienen receptores Fc ), por lo que la extirpación del bazo reduce la destrucción plaquetaria. El procedimiento es potencialmente riesgoso en los casos de PTI debido a la mayor posibilidad de sangrado significativo durante la cirugía. La remisión duradera después de la esplenectomía se logra en 60 a 80 por ciento de los casos de PTI. Aunque existe un consenso con respecto a la eficacia a corto plazo de la esplenectomía, los hallazgos sobre su eficacia a largo plazo y sus efectos secundarios son controvertidos. Después de la esplenectomía, del 11,6 al 75 por ciento de los casos de PTI recayeron y del 8,7 al 40 por ciento de los casos de PTI no respondieron a la esplenectomía. El uso de la esplenectomía para tratar la PTI ha disminuido desde el desarrollo de la terapia con esteroides y otros remedios farmacéuticos.
Transfusión de plaquetas
Normalmente, no se recomienda la transfusión de plaquetas sola, excepto en una emergencia y, por lo general, no logra producir un aumento a largo plazo del recuento de plaquetas. Esto se debe a que el mecanismo autoinmune subyacente que está destruyendo las plaquetas del paciente también destruirá las plaquetas del donante, por lo que las transfusiones de plaquetas no se consideran una opción de tratamiento a largo plazo.
Erradicación de H. pylori
En adultos, en particular en aquellos que viven en áreas con una alta prevalencia de Helicobacter pylori (que normalmente habita en la pared del estómago y se ha asociado con úlceras pépticas ), se ha demostrado que la identificación y el tratamiento de esta infección mejoran los recuentos de plaquetas en un tercio de los pacientes. En un quinto, el recuento de plaquetas se normalizó por completo; esta tasa de respuesta es similar a la encontrada en el tratamiento con rituximab, que es más caro y menos seguro. En los niños, este enfoque no está respaldado por evidencia, excepto en áreas de alta prevalencia. Las pruebas de urea en el aliento y las pruebas de antígenos en heces funcionan mejor que las pruebas serológicas ; además, la serología puede dar un falso positivo después del tratamiento con IgIV.
Otros agentes
- La dapsona (también llamada difenilsulfona, DDS o avlosulfon) es un medicamento antiinfeccioso de sulfona. La dapsona también puede ser útil para tratar el lupus, la artritis reumatoide y como tratamiento de segunda línea para la PTI. El mecanismo por el cual la dapsona ayuda en la PTI no está claro, pero se observa un aumento en el recuento de plaquetas en 40 a 60% de los receptores.
- El uso no indicado en la etiqueta de rituximab , un anticuerpo monoclonal quimérico contra el antígeno de superficie de células B CD20 , a veces puede ser una alternativa eficaz a la esplenectomía. Sin embargo, pueden ocurrir efectos secundarios significativos y los ensayos controlados aleatorios no son concluyentes.
Pronóstico
Es poco común que las personas con PTI presenten hemorragias graves (solo el 5% de las personas afectadas). Sin embargo, dentro de los cinco años posteriores al diagnóstico, el 15% de las personas afectadas son hospitalizadas por complicaciones hemorrágicas.
Epidemiología
Se considera que un recuento de plaquetas normal está en el rango de 150 000 a 450 000 por microlitro (μl) de sangre para la mayoría de las personas sanas. Por lo tanto, uno puede considerarse trombocitopénico por debajo de ese rango, aunque el umbral para un diagnóstico de PTI no está vinculado a ningún número específico.
La incidencia de PTI se estima en 50 a 100 nuevos casos por millón por año, y los niños representan la mitad de ese número. Al menos el 70 por ciento de los casos infantiles terminarán en remisión dentro de los seis meses, incluso sin tratamiento. Además, un tercio de los casos crónicos restantes generalmente remitirán durante la observación de seguimiento, y otro tercio terminará con una trombocitopenia leve (definida como un recuento de plaquetas por encima de 50.000). Se han identificado varios genes y polimorfismos relacionados con la inmunidad que influyen en la predisposición a la PTI, con el alelo FCGR3a-V158 y KIRDS2 / DL2 aumentando la susceptibilidad y KIR2DS5 demostrado ser protector.
La PTI suele ser crónica en los adultos y la probabilidad de una remisión duradera es del 20 al 40 por ciento. La proporción de hombres a mujeres en el grupo de adultos varía de 1: 1,2 a 1,7 en la mayoría de los rangos de edad (los casos infantiles son aproximadamente iguales para ambos sexos) y la edad media de los adultos en el momento del diagnóstico es de 56 a 60 años. La proporción entre casos de adultos masculinos y femeninos tiende a aumentar con la edad. En los Estados Unidos, se cree que la población crónica adulta es de aproximadamente 60.000, y las mujeres superan en número a los hombres en aproximadamente 2 a 1, lo que ha dado lugar a que la PTI se considere una enfermedad huérfana .
La tasa de mortalidad por PTI crónica varía, pero tiende a ser más alta en relación con la población general para cualquier rango de edad. En un estudio realizado en Gran Bretaña , se observó que la PTI causa una tasa de mortalidad aproximadamente un 60 por ciento más alta en comparación con los sujetos del mismo sexo y edad sin PTI. Este mayor riesgo de muerte con PTI se concentra principalmente en las personas de mediana edad y ancianos . El noventa y seis por ciento de las muertes relacionadas con la PTI reportadas fueron personas de 45 años o más. No se observaron diferencias significativas en la tasa de supervivencia entre hombres y mujeres.
El embarazo
Los autoanticuerpos antiplaquetarios en una mujer embarazada con PTI atacarán las propias plaquetas del paciente y también atravesarán la placenta y reaccionarán contra las plaquetas fetales. Por tanto, la PTI es una causa importante de trombocitopenia inmunitaria fetal y neonatal. Aproximadamente el 10% de los recién nacidos afectados por PTI tendrán recuentos de plaquetas <50.000 / uL y entre el 1% y el 2% tendrán un riesgo de hemorragia intracerebral comparable al de los bebés con trombocitopenia aloinmune neonatal (NAIT).
Ninguna prueba de laboratorio puede predecir de manera confiable si ocurrirá trombocitopenia neonatal. El riesgo de trombocitopenia neonatal aumenta con:
- Madres con antecedentes de esplenectomía por PTI
- Madres que tuvieron un bebé anterior afectado con PTI
- Recuento de plaquetas gestacional (materno) inferior a 100.000 / uL
Se recomienda que las mujeres embarazadas con trombocitopenia o un diagnóstico previo de PTI se realicen pruebas de anticuerpos antiplaquetarios séricos. Una mujer con trombocitopenia sintomática y un anticuerpo antiplaquetario identificable debe iniciar un tratamiento para su PTI, que puede incluir esteroides o IgIV. El análisis de sangre fetal para determinar el recuento de plaquetas generalmente no se realiza ya que la trombocitopenia inducida por ITP en el feto es generalmente menos grave que la NAIT. Las transfusiones de plaquetas se pueden realizar en recién nacidos, según el grado de trombocitopenia. Se recomienda realizar un seguimiento de los recién nacidos con recuentos seriados de plaquetas durante los primeros días después del nacimiento.
Historia
Después de los informes iniciales del médico portugués Amato Lusitano en 1556 y Lazarus de la Rivière (médico del rey de Francia) en 1658, fue el médico y poeta alemán Paul Gottlieb Werlhof quien en 1735 escribió el informe inicial más completo sobre la púrpura de ITP. Las plaquetas eran desconocidas en ese momento. El nombre "enfermedad de Werlhof" se usó más ampliamente antes de que el nombre descriptivo actual se hiciera más popular. Las plaquetas se describieron a principios del siglo XIX y, en la década de 1880, varios investigadores relacionaron la púrpura con anomalías en el recuento de plaquetas. El primer informe de una terapia exitosa para la PTI fue en 1916, cuando un joven estudiante de medicina polaco , Paul Kaznelson , describió la respuesta de una paciente a una esplenectomía . La esplenectomía siguió siendo un remedio de primera línea hasta la introducción de la terapia con esteroides en la década de 1950.
Referencias
enlaces externos
| Clasificación | |
|---|---|
| Recursos externos |